© Universität Bielefeld
uni.news
Published on
11. Juni 2012
Category
General
Researchers discover hereditary enzyme deficiency
The Bielefeld biochemist Thomas Dierks and his team establish the cause and develop a treatment concept
An international research team headed by Professor Dr. Thomas Dierks at Bielefeld University has discovered a hereditary enzyme deficiency. It is a subform of the mucopolysaccharidosis syndrome and has been termed MPS IIIE or – after its discoverer – ‘Dierks’s disorder’. It leads to a progressive loss of mental abilities in mice, particularly to learning and coordination difficulties along with forgetfulness. The biochemist Dierks and his team have not only identified the disorder but also developed a treatment concept. Their findings are being published this week (CW 24) in the renowned journal ‘Proceedings of the National Academy of Sciences of the USA‘ (PNAS).
 Enzymes control the constitution and degradation of nutrient and messenger substances in the human body. If the body produces a defective enzyme due to an inherited deficiency, this control breaks down. The individual becomes ill because, for example, substances can no longer be degraded and accumulate in the body.
Enzymes control the constitution and degradation of nutrient and messenger substances in the human body. If the body produces a defective enzyme due to an inherited deficiency, this control breaks down. The individual becomes ill because, for example, substances can no longer be degraded and accumulate in the body.
Dierks and his colleagues have discovered that a deficiency of the enzyme arylsulfatase G (ARSG) triggers the disease MPS IIIE in mice. Actually, the enzyme – among others – is responsible for degrading the carbohydrate heparan sulfate. This process occurs within the cells – inside the lysosomes. These ‘recycling plants’ in the cells break down no longer needed heparan sulfate molecules into their smallest components, which later are used to reassemble new molecules.
Heparan sulfate is a long-chain molecule. This chain can only be broken down from one end and only step by step. This process is performed by various enzymes including ARSG. If one of the enzymes is deficient because of a genetic defect, the complete degradation process comes to a stop. The molecular chains remain largely intact and accumulate more and more in the lysosome, which finally stops functioning. Then, also other substances such as proteins and lipids also accumulate because they are no longer degraded. The lysosome grows and grows until it damages the entire cell and eventually destroys it.
At the beginning of the study (2003), only seven of the enzymes involved in the degradation of heparan sulfate were known. Dierks and his team were searching specifically for one missing enzyme. They knew that a total of at least nine different enzymes would be needed for the complete degradation of heparan sulfate – two of these (both of them sulfatases) had still not been discovered. Every inherited deficiency in one of these enzymes corresponds to a disorder belonging to the mucopolysaccharidosis syndrome. ‘When we started the study, we suspected that arylsulfatase G was involved in the degradation of heparan sulfate’, says Dierks. To test this assumption, his team bred mice in which arylsulfatase G was deficient. Their suspicion was confirmed: With increasing age, the mice revealed high concentrations of heparan sulfate in the brain, the liver, and the kidneys. Professor Jeffrey D. Esko from the University of California San Diego (USA) is also working in this project. He and his team analysed tissue samples with a mass spectrometer and confirmed the finding that the blocked cleavage of a specific sulfate group is the cause of the accumulation of heparan sulfate.
Using behaviour tests, Dierks's team found that mice with an ARSG deficiency suffer from cognitive problems once they reach the age of 12 months. If they enter an open field, they stick to the safer periphery and, unlike healthy littermates, do not have the courage to explore the centre. They also fail to master a water maze. For a long time, the mice had been trained successfully to swim through a pool filled with a milky liquid until they found a platform hidden beneath its surface. However, as soon as they were 12 months old, the mice failed to memorize where the platform was located. They took much longer than before to discover it. Younger and healthy mice had no problems in finding the platform again. The defect is in the brain. In a study of tissue samples from the animals’ cerebellum, the research team showed that through the accumulation of heparan sulfate, the Purkinje cells in the cerebellum die off and – accompanied by inflammation – are replaced by new cells. However, according to Dierks, these glia cells have only a supporting function and do not form any new connections to nerve cells.
A major success for the researchers is that their findings can be used to develop a treatment for this hereditary disorder and test this treatment on ARSG deficient mice. They are producing ARSG enzyme in a biotechnology process with the help of genetically modified cell cultures. In sick mice that are regularly injected with a solution containing the enzyme, further damage to their organs should be stopped, Dierks believes. Similar treatments are successfully applied to patients with other mucopolysaccharidosis disorders. ‘The biochemical processes underlying such lysosomal storage disorders are principally the same in all mammals. However, effects are more severe in human beings because of their greater life span’, says Dierks. An early diagnosis of these disorders can be difficult because the onset is frequently insidious. Sometimes, the disorder can only be recognized through its symptoms in adolescence. And then the diagnosis is difficult for a physician, Thomas Dierks says, ‘because systematic screenings are performed only in childhood, and one does not initially consider a genetic cause. However, treatment has to start as early as possible’. ‘Dierks's disorder’ can be diagnosed unequivocally with a mass spectrometer – the researchers have developed their own procedure for this.
Alongside scientists at Bielefeld University, Thomas Dierks’s research team includes colleagues at the Katholieke Universiteit Leuven, Belgium; the University of California San Diego, USA, the University of Göttingen, and the University of Kiel.
Original publication:
Arylsulfatase G Inactivation Causes Loss of Heparan Sulfate 3-O-Sulfatase Activity and Mucopolysaccharidosis in Mice, Björn Kowalewski, William C. Lamanna, Roger Lawrence, Markus Damme, Stijn Stroobants, Michael Padva, Ina Kalus, Marc-André Frese, Torben Lübke, Renate Lüllmann-Rauch, Rudi D’Hooge, Jeffrey D. Esko, Thomas Dierks, Proceedings of the National Academy of Sciences of the USA, June 2012, dx.doi.org/10.1073/pnas.1202071109.
Contact:
Prof. Dr. Thomas Dierks, Bielefeld University
Faculty of Chemistry, Research Group Biochemistry I
Telephone: 0521 106-2092
Email: thomas.dierks@uni-bielefeld.de

An international research team headed by Professor Dr. Thomas Dierks at Bielefeld University has discovered a hereditary enzyme deficiency. It is a subform of the mucopolysaccharidosis syndrome and has been termed MPS IIIE or – after its discoverer – ‘Dierks’s disorder’. It leads to a progressive loss of mental abilities in mice, particularly to learning and coordination difficulties along with forgetfulness. The biochemist Dierks and his team have not only identified the disorder but also developed a treatment concept. Their findings are being published this week (CW 24) in the renowned journal ‘Proceedings of the National Academy of Sciences of the USA‘ (PNAS).
The team of Scientists (left to right): Dr. Markus Damme and Björn Kowalewski (in the front), Professor Dr. Thomas Dierks and Apl. Professor Dr. Torben Lübke (background).
Dierks and his colleagues have discovered that a deficiency of the enzyme arylsulfatase G (ARSG) triggers the disease MPS IIIE in mice. Actually, the enzyme – among others – is responsible for degrading the carbohydrate heparan sulfate. This process occurs within the cells – inside the lysosomes. These ‘recycling plants’ in the cells break down no longer needed heparan sulfate molecules into their smallest components, which later are used to reassemble new molecules.
Heparan sulfate is a long-chain molecule. This chain can only be broken down from one end and only step by step. This process is performed by various enzymes including ARSG. If one of the enzymes is deficient because of a genetic defect, the complete degradation process comes to a stop. The molecular chains remain largely intact and accumulate more and more in the lysosome, which finally stops functioning. Then, also other substances such as proteins and lipids also accumulate because they are no longer degraded. The lysosome grows and grows until it damages the entire cell and eventually destroys it.
At the beginning of the study (2003), only seven of the enzymes involved in the degradation of heparan sulfate were known. Dierks and his team were searching specifically for one missing enzyme. They knew that a total of at least nine different enzymes would be needed for the complete degradation of heparan sulfate – two of these (both of them sulfatases) had still not been discovered. Every inherited deficiency in one of these enzymes corresponds to a disorder belonging to the mucopolysaccharidosis syndrome. ‘When we started the study, we suspected that arylsulfatase G was involved in the degradation of heparan sulfate’, says Dierks. To test this assumption, his team bred mice in which arylsulfatase G was deficient. Their suspicion was confirmed: With increasing age, the mice revealed high concentrations of heparan sulfate in the brain, the liver, and the kidneys. Professor Jeffrey D. Esko from the University of California San Diego (USA) is also working in this project. He and his team analysed tissue samples with a mass spectrometer and confirmed the finding that the blocked cleavage of a specific sulfate group is the cause of the accumulation of heparan sulfate.
Using behaviour tests, Dierks's team found that mice with an ARSG deficiency suffer from cognitive problems once they reach the age of 12 months. If they enter an open field, they stick to the safer periphery and, unlike healthy littermates, do not have the courage to explore the centre. They also fail to master a water maze. For a long time, the mice had been trained successfully to swim through a pool filled with a milky liquid until they found a platform hidden beneath its surface. However, as soon as they were 12 months old, the mice failed to memorize where the platform was located. They took much longer than before to discover it. Younger and healthy mice had no problems in finding the platform again. The defect is in the brain. In a study of tissue samples from the animals’ cerebellum, the research team showed that through the accumulation of heparan sulfate, the Purkinje cells in the cerebellum die off and – accompanied by inflammation – are replaced by new cells. However, according to Dierks, these glia cells have only a supporting function and do not form any new connections to nerve cells.
A major success for the researchers is that their findings can be used to develop a treatment for this hereditary disorder and test this treatment on ARSG deficient mice. They are producing ARSG enzyme in a biotechnology process with the help of genetically modified cell cultures. In sick mice that are regularly injected with a solution containing the enzyme, further damage to their organs should be stopped, Dierks believes. Similar treatments are successfully applied to patients with other mucopolysaccharidosis disorders. ‘The biochemical processes underlying such lysosomal storage disorders are principally the same in all mammals. However, effects are more severe in human beings because of their greater life span’, says Dierks. An early diagnosis of these disorders can be difficult because the onset is frequently insidious. Sometimes, the disorder can only be recognized through its symptoms in adolescence. And then the diagnosis is difficult for a physician, Thomas Dierks says, ‘because systematic screenings are performed only in childhood, and one does not initially consider a genetic cause. However, treatment has to start as early as possible’. ‘Dierks's disorder’ can be diagnosed unequivocally with a mass spectrometer – the researchers have developed their own procedure for this.
Alongside scientists at Bielefeld University, Thomas Dierks’s research team includes colleagues at the Katholieke Universiteit Leuven, Belgium; the University of California San Diego, USA, the University of Göttingen, and the University of Kiel.
Original publication:
Arylsulfatase G Inactivation Causes Loss of Heparan Sulfate 3-O-Sulfatase Activity and Mucopolysaccharidosis in Mice, Björn Kowalewski, William C. Lamanna, Roger Lawrence, Markus Damme, Stijn Stroobants, Michael Padva, Ina Kalus, Marc-André Frese, Torben Lübke, Renate Lüllmann-Rauch, Rudi D’Hooge, Jeffrey D. Esko, Thomas Dierks, Proceedings of the National Academy of Sciences of the USA, June 2012, dx.doi.org/10.1073/pnas.1202071109.
Contact:
Prof. Dr. Thomas Dierks, Bielefeld University
Faculty of Chemistry, Research Group Biochemistry I
Telephone: 0521 106-2092
Email: thomas.dierks@uni-bielefeld.de
Professor Dr. Thomas Dierks and his international team have verified the disorder ‘mucopolysaccharidosis IIIE’ and its molecular basis. Now they are studying how to stop it.
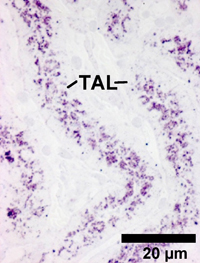
Onset of ‘Dierks’s disorder’: The microscopic photo shows strong accumulations of heparan sulfate (blue-red colouring) in cells of the thick ascending limb (TAL) of renal tubules. The cause is ARSG enzyme deficiency.